
Nou Reglament de productes sanitaris
Autor: TIC Salut Social / 9 de març de 2020

9 de març de 2020
El nou Reglament de productes sanitaris està a punt de començar a aplicar-se. Acabarà així un període de transició que va començar l’any 2017 amb la publicació dels Reglaments (EU) 2017/745 (MDR1) i (EU) 2017/746 (IVDR2) (aquest últim per a productes sanitaris de diagnòstic in vitro, que començarà a aplicar-se el 26 de maig de 2022).
El pròxim 26 de maig 2020 el Reglament (EU) 2017/745 començarà la seva aplicació, obligatòriament, en els següents productes sanitaris:
- Productes de Classe I sota el MDD3, que continuaran sent de Classe I sota el MDR i, per tant, no necessiten involucrar a un organisme notificat.
- Productes implementables a mida de Classe III.
- Productes que utilitzen derivats de teixits humans.
- Productes combinats (aquells que inclouen una part de medicament i una altra de producte sanitari) sota l’Article 117.
Com a excepció, 6 mesos a partir de la data d’adopció de l’especificació comuna que publiqui la comissió, en lloc del 26 de maig de 2020 per a:
- Productes de l’Annex XVI sense finalitats mèdiques, com ara làsers, equips de liposucció, etc.
A què es deuen aquests canvis?
El Reglament reemplaçarà a les Directives 93/42/EEC (MDD) (de producte sanitari) i 90/385/EEC (AIMDD) (de producte sanitari implementable actiu) fins ara vigents.
Què buscava la Comissió Europea amb aquest gran canvi? Diverses coses, però entre elles la primera i més important millorar la seguretat dels pacients i la Salut Pública.
Els nous Reglaments suposen un salt qualitatiu i quantitatiu important.
En primer lloc, són Reglaments que entren en vigor en tots els països de la Unió Europea, mentre que les Directives vigents, tenen en cada país la seva transposició nacional, que donen cabuda a diferents interpretacions de la normativa aplicable.
En el cas d’Espanya tenim els Reials decrets RD 1616/2009 per als productes sanitaris implementables actius i l’RD 1591/2009 per als productes sanitaris. Tots dos afegeixen requisits a les Directives vigents.
Figura 1. Reglaments i Directives
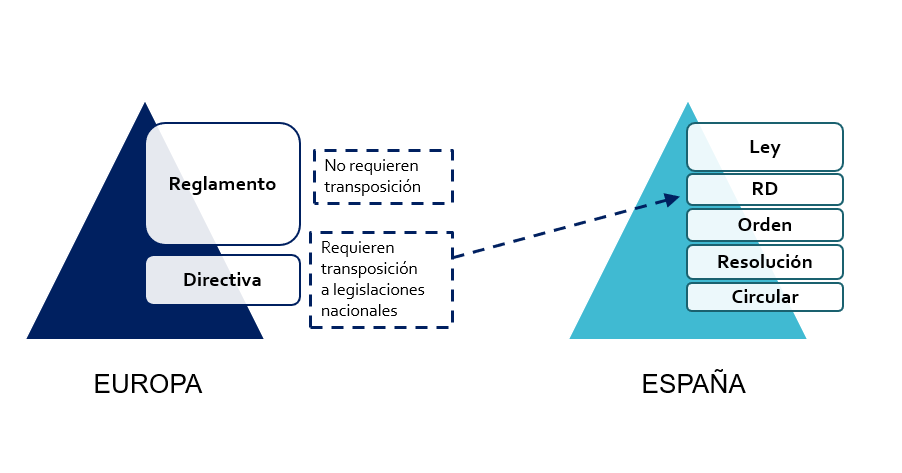
Font: CSV Experts
Figura 2: Legislació vigent a Europa

Font: CSV Experts
En segon lloc, estem parlant de Directives dels anys 90, 1990 per a la Directiva de producte sanitari implementable actiu i 1993 per a la Directiva de producte sanitari. Ambdues necessitaven actualitzar-se per a aconseguir adaptar les exigències del sector als importants avenços que s’han donat a nivell científic i tecnològic.
D’acord amb les dades publicades per l’associació MedTech Europe la tecnologia mèdica es caracteritza per una constant innovació, resultat d’una I+D propi d’aquesta indústria. Els productes sanitaris solen necessitar entre 17-24 mesos per arribar al mercat. Les següents xifres ens poden donar una idea: l’any 2017, l’oficina de registre de patents va registrar més de 13.000 peticions de patents de tecnologia mèdica (7,9% del total de peticions), el 40% d’aquestes peticions provenien de països europeus (EU28, Noruega i Suïssa), i el 60% restant d’altres països en la seva majoria dels Estats Units (37%). Hi ha a tota Europa unes 27.000 empreses de tecnologia mèdica de les quals una immensa majoria (95%) són petites i mitjanes empreses, i dins d’elles la majoria empren a menys de 50 persones (empreses petites i microempreses).
Però sens dubte, el punt crític que va marcar la necessitat de realitzar canvis a nivell regulador i augmentar els controls va ser el frau de l’empresa francesa PIP (Poly Implant Prothèse), en el qual es va descobrir que un dels majors fabricants d’implants mamaris del món estava fabricant implants amb silicona industrial no quirúrgica, després d’aparèixer nombrosos casos de trencaments, i estar implicats en algunes morts a causa de càncers. S’estima, sobre la base de dades facilitades per l’Agència de Productes de Salut de França (ANSM), que hi ha almenys unes 400.000 persones afectades en 65 països, unes 7.500 trencaments detectats i unes 3.000 dones que han informat d’efectes no desitjats, la majoria inflamació en els seus implants. L’empresa es va declarar en fallida i al tancar en 2010, amb moltes persones esperant rebre una indemnització per danys i perjudicis. A partir d’aquest escàndol la comissió europea va crear l’anomenat pla d’acció PIP, que va suposar les bases del que són els nous Reglaments, amb especial atenció als següents punts:
1. Revisar el funcionament dels organismes notificats.
2. Augment del Seguiment – vigilància postcomercialització.
3. Coordinació d’organismes notificats i autoritats competents en el cas de vigilància.
4. Comunicació i transparència.
Què és el que ha canviat?
L’impacte per a tots els agents econòmics, sobretot per a fabricants i organismes notificats és considerable ja que han augmentat els requisits que apliquen a cadascun:
Ha canviat la classificació, a més estricta, de nombrosos productes sanitaris, la qual cosa implica majors requisits a nivell regulador.
Es requereixen major nombre de dades clíniques i s’esperen més recerques clíniques, sobretot per als productes de major risc (Classe IIb i Classe III).
Majors requisits de seguiment postcomercialització. S’exigeix a les organitzacions que siguin molt proactives en la recopilació de dades després de la comercialització dels productes o serveis.
S’ha creat la Base de dades EUDAMED (European Database on Medical Devices). Es tracta d’un projecte molt ambiciós, que pretén centralitzar el seguiment de tots els productes sanitaris, des de la seva introducció al mercat fins a la vigilància o els estudis clínics. EUDAMED disposarà una part oberta al públic perquè qualsevol profà la pugui consultar.
Necessitat de UDI (Unique Device Identification): la traçabilitat és un dels cavalls de batalla dels nous Reglaments i cada producte sanitari ha de tenir un número d’identificació única.
Targeta per a implants: els implants tenen requisits més estrictes de traçabilitat, buscant l’eficàcia en cas d’alertes, o retirades de producte. La targeta serà lliurada al pacient, sent per tant coneixedor del fabricant de l’implant i dades de traçabilitat específics.
Suficient cobertura d’assegurança en les empreses per a cobrir denúncies dels danys potencials que pugui causar.
S’inclou la figura de persona responsable de compliment de la normativa dins de les organitzacions fabricadores i dels representants autoritzats.
El Reglament afecta també a productes amb finalitat cosmètica que tenen el mateix mecanisme d’acció i per tant riscos, que productes sanitaris equivalents, com per exemple làsers o equips de liposucció.
Els organismes notificats han de passar un procés de recertificación i se’ls sotmetrà a un seguiment estricte, així com a auditories no anunciades.
Aquest aspecte, encara que figuri com l’últim de la llista, és d’especial importància com ja bé sap el sector. Segons les Directives hi havia 58 organismes notificats designats. En el moment de la redacció d’aquest article, principis de març 2020, tenim només 11 organismes notificats designats per a Reglament 2017/745 (EU). Aquest aspecte és de crucial importància atès que ara més que mai l’elecció d’organisme notificat és una decisió molt estratègica per a l’organització.
Figura 3: Els nous Reglaments

Font: EC.
Figura 4: Elements a considerar

Font: EC.
El nou Reglament també apunta a tots els agents implicats en l’arribada al mercat d’un producte o servei sanitari (fabricant, representant autoritzat, importador o distribuïdor). Són ells també, garants de la conformitat amb la normativa, la traçabilitat i la vigilància.
Figura 5. Noves característiques

Font: EC.
L’impacte a les organitzacions és per això considerable, implicant l’augment de recursos destinats a assegurar la conformitat amb la nova regulació. Per a assegurar una transició més suau, i no desatendre el mercat, el Reglament entrarà en vigor gradualment (veure figura 6). Aquells productes que necessitin, per la seva tipologia de risc, un organisme notificat per a obtenir el seu marcatge CE, podran tenir els seus productes al mercat mentre tinguin un certificat CE vigent contra MDD, com a màxim fins a l’any 2024, i un any més, 2025 en distribució.
Això sí, els productes que continuïn al mercat segons MDD no poden tenir modificacions de disseny durant aquest període i han de complir igualment amb les obligacions d’UDI, registre d’agents econòmics a EUDAMED així que entri en vigor i seguiment postcomercialiyzació segons Reglament MDR.
Figura 6. Calendari de transició de las Directives als Reglaments

Font: EC.
Alguns reptes
La disponibilitat d’organismes notificats és un clar coll d’ampolla, tal com s’ha comentat anteriorment. Per això, disposar d’un certificat CE vàlid segons les Directives MDD o AIMDD pot ser un avantatge per a continuar l’adaptació a MDR.
EUDAMED ha retrassat la seva entrada en vigor fins al 26 de maig de 2022, per a coincidir amb la data d’aplicació d’IVDR. La base de dades és un projecte molt ambiciós i complex, que genera encara molts dubtes sobre el seu funcionament en tots els mòduls, especialment els de seguiment postcomercialització, vigilància i recerques clíniques.
El tenir dades clíniques suficients i de qualitat és un clar repte per a les empreses. Suposarà una important necessitat d’inversió en recursos, així com un impacte econòmic sobretot en les petites i mitjanes empreses.
Conclusions: Com ens preparem?
Així doncs, com a fabricant, quins són els punts més importants que cobrir per a continuar venent els nostres productes i serveis sanitaris a l’UE i no perdre el marcatge CE?
- Assegurar-nos de la classificació dels productes sanitaris que comercialitzem i generar un informe de classificació segons l’annex VIII de MDR.
- Dur a terme una anàlisi de desviacions on evidenciem quina documentació necessitem per a estar de conformitat amb MDR.
- Assegurar la disponibilitat de suficients dades clíniques per a justificar la seguretat i eficàcia dels nostres productes. I disposar d’un informe d’avaluació clínica d’acord amb MDR.
- Implantar en les organitzacions un eficaç sistema de seguiment poscomercialización amb cerca proactiva de dades.
- Disposar d’una assegurança de responsabilitat civil que cobreixi indemnitzacions en productes o serveis defectuosos que afectin la salut dels pacients.
- Disposar d’UDI i registrar a EUDAMED quan estigui disponible, previsiblement al maig de 2022.
_____________________
- MDR: Medical Device Regulations
- IVDR: In Vitro Diagnostic Regulations
- MDD: Medical Device Directives
_____________________
Referencies
- European Commission, (2020). Internal Market, Industry, Entrepreneurship and SMEs. Recuperat el 9 de març de 2020 de https://ec.europa.eu/growth/sectors/medical-devices_en
- Agencia Española de Medicamentos y Productos Sanitarios, (2020). Productos sanitarios. Recuperat el 9 de març de 2020 de https://www.aemps.gob.es/productos-sanitarios/productossanitarios_prodsanitarios/
- Medtech Europe, (2020). New medical technology. Recuperat el 9 de març de 2020 de https://www.medtecheurope.org/new-medical-technology-regulations/
-
El Reglament reemplaçarà a les Directives 93/42/EEC (MDD) (de producte sanitari) i 90/385/EEC (AIMDD) (de producte sanitari implantable actiu) fins ara vigents